Produits depuis 20 ans, les biosimilaires sont encore peu connus du grand public. Afin d’accompagner au mieux leurs patients, les infirmières ont besoin de bien maîtriser leurs caractéristiques.
Alors que la dispensation des médicaments génériques, au bout de 30 ans de campagne de communication, se banalise, la mise à disposition de médicaments biosimilaires ainsi que le dispositif d’interchangeabilité et de substitution de ces produits relancent les débats. Afin de pouvoir répondre aux interrogations des patients, l’infirmière a besoin de bien connaître les particularités de ces médicaments un peu à part. Sachant que, comme pour les génériques, le principal frein à l’adoption des médicaments biosimilaires est qu’ils ne présentent pas un gain direct et immédiat pour le patient. Moins chers que les autres médicaments, leur intérêt est d’abord économique. Ils doivent cependant permettre à terme d’optimiser la qualité de la prise en charge des patients.
QU’EST-CE QU’UN MÉDICAMENT BIOLOGIQUE ?
On parle de médicament biologique par opposition aux médicaments « classiques » obtenus par synthèse chimique. Aussi appelé biomédicament, il est issu d’une synthèse biologique ayant comme point de départ une source biologique telle que des cellules ou des organismes vivants. Actuellement, la plupart des biomédicaments utilisés en médecine contiennent des substances actives composées de protéines plus ou moins grandes et de structure plus ou moins complexe, comme les hormones (insulines, érythropoïétine, hormones de croissance), les facteurs de coagulation ou les anticorps monoclonaux.
QU’EST-CE QU’UN BIOSIMILAIRE ?
Selon l’article L.5121-1 15 du code de la santé publique, « un médicament biologique similaire est un médicament biologique de même composition qualitative et quantitative en substance active et de même forme pharmaceutique qu’un médicament biologique de référence mais qui ne remplit pas les conditions pour être regardé comme une spécialité générique en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication et nécessitant que soient produites des données précliniques et cliniques supplémentaires dans des conditions déterminées par voie réglementaire ». Autrement dit, lorsqu’un brevet d’exploitation tombe dans le domaine public, les laboratoires pharmaceutiques peuvent commercialiser une « copie » d’un biomédicament, ce dernier devenant médicament de référence.
Le biosimilaire doit avoir la même substance active, le même dosage et une forme pharmaceutique identique à celle du médicament de référence. Néanmoins, la variabilité naturelle de la source biologique et du procédé de fabrication propre à chaque fabricant peut être à l’origine de légères différences entre le médicament biosimilaire et son médicament de référence. Cette variabilité ne doit pas avoir d’impact sur l’efficacité, la sécurité d’utilisation ni la tolérance au traitement.
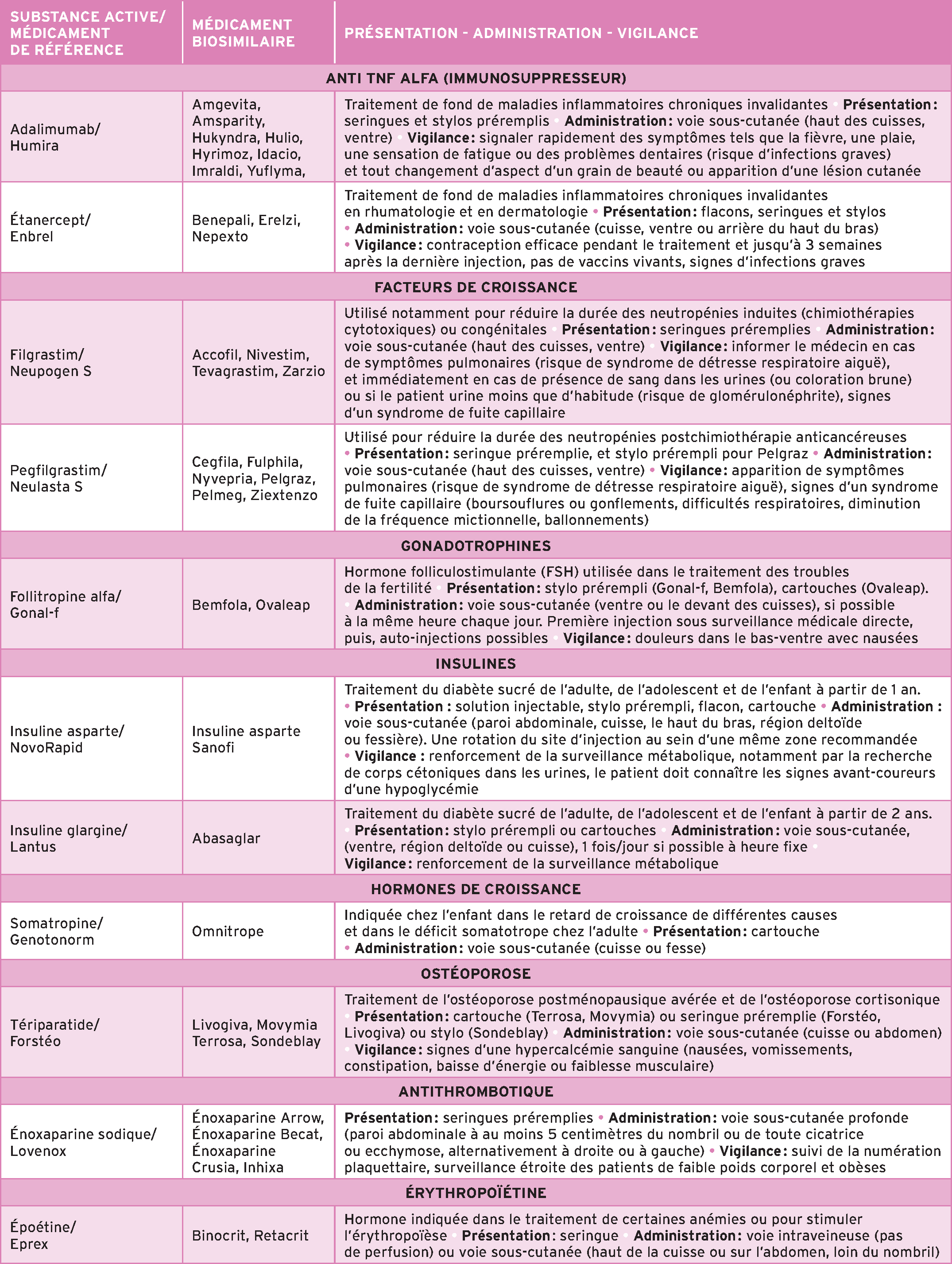
Les premiers biosimilaires à avoir obtenu une autorisation européenne ont été ceux de l’hormone de croissance humaine en 2006. Depuis, le marché des médicaments biosimilaires se développe rapidement, au fur et à mesure de l’expiration des brevets des biomédicaments. À ce jour, l’Agence nationale de sécurité du médicament (ANSM) répertorie 16 groupes de biosimilaires dont 12 sont disponibles en pharmacie d’officine (voir tableau p. 32).
QUEL CADRE RÉGLEMENTAIRE ?
Autorisation de mise sur le marché. Un cadre réglementaire spécifique aux médicaments bio-similaires a été établi en Europe dès 2005. Toute demande d’autorisation de mise sur le marché (AMM) dans les pays de l’Union européenne est ainsi soumise à une procédure d’examen centralisée par l’Agence européenne du médicament (EMA). L’évaluation par l’EMA donne lieu à un avis scientifique envoyé à la Commission européenne, qui accorde une autorisation de mise sur le marché valable dans l’ensemble de l’Union européenne (UE).
Concept de biosimilarité : il repose sur le principe essentiel de la comparaison de deux médicaments issus de la biotechnologie, le médicament de référence (autorisé depuis plus de huit ans dans l’UE) et le médicament qui souhaite être déclaré biosimilaire. L’évaluation diffère sensiblement de celle des médicaments génériques qui ne requiert qu’un dossier de qualité et l’étude de bioéquivalence. En effet, la comparaison des biosimilaires porte, elle, sur une analyse des propriétés physicochimiques (qualité), pharmacodynamiques, toxicologiques (sécurité) et cliniques (efficacité et tolérance). Ces études comparatives n’ont pas pour but d’établir le profil de sécurité du médicament, car celui-ci est connu avec le médicament de référence, mais d’identifier d’éventuelles différences de profil pharmacologique qui pourraient avoir un effet sur le profil d’efficacité clinique.
QUELLES GARANTIES EN TERMES DE SÉCURITÉ ?
Études d’immunogénicité. Les médicaments biologiques (qu’il s’agisse des médicaments de référence ou des biosimilaires) peuvent provoquer une réponse immunitaire non désirée chez les patients traités, conduisant à la production d’anti-corps antimédicaments. On parle d’immunogénicité. Le profil d’immunogénicité d’une molécule est systématiquement étudié car elle peut être à l’origine d’effets indésirables graves (anaphylaxie, hypersensibilité retardée) ou conduire à une diminution de l’efficacité du traitement. Dans la pratique, les tests cliniques ont démontré que les réactions graves liées à une augmentation de la réponse immunitaire sont très rares et que dans la majorité des cas une réponse immunitaire inadaptée n’induit pas de conséquence clinique. L’expérience montre même qu’au cours d’un traitement au long cours, un patient est susceptible de recevoir des médicaments biologiques présentant de légères différences (inhérentes à la source biologique ou à une modification du procédé de fabrication) sans que cela n’induise de réponse immunitaire nocive. Ainsi, rien ne laisse croire qu’une réponse immunitaire nocive puisse être déclenchée par la permutation d’un médicament de référence avec son biosimilaire.
Néanmoins, les études d’immunogénicité clinique et des études comparatives avec le biomédicament de référence sont requises pour l’approbation des biosimilaires par l’EMA.
Extrapolation. Certains biosimilaires peuvent avoir moins d’indications que le médicament de référence auquel ils sont liés. Mais quand un médicament biosimilaire démontre une forte similarité avec un biomédicament de référence et qu’il affiche une sécurité et une efficacité comparables pour une indication thérapeutique donnée, une extrapolation pour les autres indications de l’AMM du biomédicament de référence est possible, même s’il s’agit de domaines thérapeutiques différents. Cette particularité réglementaire permet de limiter les essais cliniques pour certaines indications, mais cette tolérance n’est pas systématiquement appliquée et des études complémentaires peuvent être demandées.
Surveillance. Les médicaments biosimilaires, comme tous les biomédicaments, sont soumis à une surveillance renforcée qui permet aux autorités sanitaires de disposer de données complémentaires après la commercialisation. Cette surveillance est signalée par la présence d’un triangle noir inversé sur les notices et sur les résumés des caractéristiques de chaque spécialité.
QU’EST-CE QUE L’INTERCHANGEABILITÉ ?
L’interchangeabilité est un acte médical qui consiste, à l’initiative du prescripteur, à remplacer un médicament biologique (médicament de référence ou biosimilaire) par un autre médicament similaire. Contrairement aux dossiers d’AMM, les règles autorisant ou non l’interchangeabilité entre biomédicaments similaires ne dépendent pas d’une décision de l’EMA. Chaque État membre de l’Union européenne peut légiférer et voter ses propres lois. En France, l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) avait initialement recommandé de ne pas remplacer un biomédicament par un autre en cours de traitement. Cependant, compte tenu de l’évolution des connaissances et des analyses des données d’efficacité et de sécurité des biomédicaments, l’agence est revenue sur ses recommandations en mai 2016 et la loi de financement de la Sécurité sociale pour 2017 a officiellement légalisé l’interchangeabilité des médicaments biologiques similaires par le médecin à tout moment du traitement (article L. 5125-23-2 du code de la santé publique).
Si l’interchangeabilité est désormais autorisée, elle doit néanmoins être raisonnée et tenir compte de l’intérêt du patient. Elle reste également conditionnée au respect de trois conditions :
→ le patient doit être informé et doit donner son accord ;
→ une surveillance clinique appropriée doit être mise en place pendant le traitement ;
→ la traçabilité des produits concernés doit être assurée (le produit doit être inscrit dans le dossier du patient).
Depuis le 1er janvier 2022 (avenant 9 à la convention médicale), un dispositif d’intéressement des médecins libéraux à la prescription des médicaments biosimilaires est entré en vigueur pour six molécules : adalimumab (Humira), énoxaparine (Lovenox), étanercept (Enbrel), follitropine alfa (Gonal-f), insuline asparte (Novorapid) et tériparatide (Forsteo).
QUELLES SONT LES RÈGLES DE PRESCRIPTION ET DE DISPENSATION ?
La prescription médicale d’un médicament biologique (médicament de référence ou biosimilaire) doit s’effectuer en dénomination commune ainsi qu’en nom de marque ou de fantaisie. Ce qui implique que si la prescription n’a indiqué que la dénomination commune internationale (DCI) sur l’ordonnance, le pharmacien ne peut pas la délivrer et doit contacter le médecin pour faire préciser le nom de marque du produit.
La substitution d’un médicament biologique par le pharmacien, inscrite dans la loi de financement de la Sécurité sociale 2022 (article 64), est entrée en application en octobre 2022. Le pharmacien peut remplacer un biomédicament par son biosimilaire, à condition que :
→ le médicament biologique similaire délivré appartienne au même groupe biologique similaire que le médicament biologique prescrit ;
→ le groupe biologique similaire figure sur une liste fixée par un arrêté conjoint des ministres chargés de la Santé et de la Sécurité sociale pris après avis de l’ANSM ;
→ le prescripteur n’ait pas exclu la possibilité de substitution par une mention expresse et justifiée portée sur l’ordonnance (selon la situation médicale du patient) ;
→ le pharmacien inscrive le nom du médicament délivré sur l’ordonnance et informe le prescripteur de la substitution.
Afin de garantir la sécurité de dispensation et d’utilisation des médicaments biologiques, l’adhésion des patients et l’ensemble des professionnels de santé aux conditions et aux contraintes liées à la substitution, l’ANSM a proposé une mise en place progressive de la substitution en ciblant, dans un premier temps, un nombre limité de médicaments biologiques substituables. Elle se limite donc pour l’heure au filgrastim (médicament de référence Neupogen) et au pegfilgrastim (médicament de référence Neulasta).
* Source : Le Moniteur des pharmacies, n° 3422, Cahier 2 Formation, du 18 juin 2022.
L’autrice déclare ne pas avoir de liens d’intérêt.
SUPPORTS DE COMMUNICATION
Pour faire connaître les médicaments biosimilaires ou accompagner une discussion avec le patient, il peut être utile de lui signaler deux documents accessibles sur ameli.fr (ou via ce raccourci : bit.ly/3TzbsBt) :
- un mémo destiné au public élaboré par l’Assurance maladie en partenariat avec l’Observatoire des médicaments dispositifs médicaux innovations thérapeutiques Centre-Val de Loire (OMéDIT Centre-Val de Loire) reprend les principales données sur les médicaments biosimilaires ;
- un guide intitulé Que dois-je savoir sur les médicaments biosimilaires ? ainsi qu’une vidéo explicative sur les biosimilaires en Europe produit par l’Agence européenne des médicaments et la Commission européenne.
Patient +
• Quel recul a-t-on sur les biomédicaments ? Les biomédicaments sont produits depuis plus de 20 ans. Il existe environ 250 produits issus des biotechnologies et 250 millions de patients ont été traités par ces produits. Ils sont utilisés avec succès dans le domaine de la prévention (vaccins) ou le traitement de maladies, comme en cancérologie, en rhumatologie et pour les maladies métaboliques.
• Quel intérêt ai-je à prendre un biosimilaire ? Les biosimilaires ne présentent pas d’intérêt thérapeutique individuel immédiat pour le patient. Si le patient est bien stabilisé avec son traitement, il est possible que le passage à un biomédicament nécessite une surveillance attentive dans les premiers temps et une réappropriation du traitement. En revanche, le fait que plusieurs laboratoires proposent des biosimilaires contribue à élargir les possibilités de ravitaillement pour pallier les ruptures d’approvisionnement d’un produit et faciliter l’accès au soin. Le biosimilaire coûte moins cher que le médicament de référence, ce qui participe à l’équilibre économique du système de santé. Les économies générées permettent d’investir dans la recherche, de développer et de financer de nouveaux médicaments. Le coût réduit des biosimilaires pourrait également permettre d’utiliser ces médicaments dans les formes plus précoces des maladies et peut-être moins sévères.
• Qu’est-ce que la prise d’un biosimilaire change pour moi ? La prise d’un biosimilaire ne change pas grand-chose au traitement habituel. Les bénéfices attendus et les risques sont identiques, les doses et le mode d’administration ainsi que la fréquence de prise sont les mêmes, le suivi du traitement est identique. Il existe parfois quelques particularités de conservation ou d’utilisation du dispositif médical associé au médicament, qui seront précisées et expliquées par les professionnels de santé au moment de la dispensation ou de la prescription.
Alexandra Blanc
Médicaments biosimilaires disponibles en pharmacie de ville